研究紹介
私たちの研究室では、アルツハイマー病を主なターゲットとし、これまで、
- 老人斑を作らない遺伝子変異(Osaka変異)の発見とその遺伝様式(劣性遺伝)の機序解明
- 新しいタウモノクローナル抗体の開発、抗生物質リファンピシンの認知症予防薬としてのドラッグ・リポジショニング、天然物由来の新しい認知症予防食品の開発
- Aβの生理作用の解明
などの研究を進めてきました。現在は、予防薬・治療薬の開発に加え、新しい診断法の開発も行っています。
アルツハイマー病とは
あるタンパク質が構造変化を起こして凝集し、脳に蓄積して、神経変性を起こす。それが原因で発症する認知症を変性性認知症と呼びます。代表的なものとしては、アルツハイマー病、前頭側頭型認知症、レビー小体型認知症などがあります。このうち最も患者数が多いのがアルツハイマー病で、認知症全体の60-70%を占めています。
認知症は脳の神経ネットワークが破壊されることで発症すると考えられています。神経ネットワークは神経細胞どうしが神経伝達物質を介して情報のやり取りをすることで成り立っており、この情報のやり取りが行われる神経細胞と神経細胞の接合部をシナプスと呼びます。シナプスの働きに障害が起こると、学習や記憶に影響が出ます。シナプス部位だけでなく神経細胞そのものが障害を受け、細胞死を起こして脱落し始めると、もの忘れ(軽度認知障害、MCI)を自覚するようになります。やがて、脳が萎縮するほどに多くの神経細胞が脱落すると、通常の生活にも支障をきたすようになって、認知症と診断されることになります。言いかえると、認知症と診断される頃にはすでに多くの神経細胞が死んでいます。そして、死んでしまった神経細胞は元に戻りません。神経細胞が死に始める前に異常を発見し、予防的な治療を始めることが大切なのです。
では、一体何がシナプスの働きを阻害し、神経細胞を死に至らしめるのでしょうか。その原因解明の鍵となるのが、脳の病理変化です。アルツハイマー病の脳では、老人斑、神経原線維変化、神経細胞死(それによる脳の萎縮)という3つの特徴的な病理変化が起こります。老人斑はアミロイドβ(以下Aβと略します)と呼ばれる40~43アミノ酸からなる小さなペプチドが互いに集まって、水に溶けない線維状の凝集体(「アミロイド線維」や「フィブリル」と呼ばれます)を形成し、細胞外に沈着したものです。一方、神経原線維変化はタウと呼ばれる352~441アミノ酸からなるタンパク質が互いに集まって、これも不溶性の線維状凝集体を形成し、神経細胞内に溜まったものです。タウは、細胞内で物質輸送を担う微小管というレールを補強するまくら木のような働きをしています。タウ分子が「リン酸化」という修飾を過剰に受けると、タウは微小管に結合できなくなって凝集し、微小管は不安定になって細胞の機能が低下すると考えられています。病理変化のうち最も早く脳に出現するのは老人斑で、次に神経原線維変化、続いて脳の萎縮(神経細胞死)が起こり、認知症が発症するのはそのあとです。老人斑形成から神経原線維変化出現までは約10年、認知症発症までは20年以上もの歳月がかかります(図1)。Aβが凝集・沈着することで老人斑ができ、老人斑が引き金となって神経原線維変化ができ、神経原線維変化が原因で神経細胞が死に、神経細胞が死ぬことによって認知症が発症する。この考えは「アミロイドカスケード仮説」(または短く「アミロイド仮説」)と呼ばれ、1992年に提唱されて以来多くの研究者に支持されてきました。
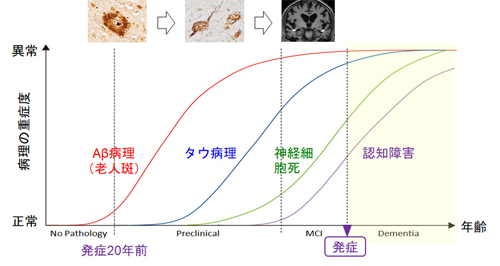
図1.アルツハイマー病の脳病理の出現から発症まで
アミロイド仮説に基づいて、これまで、Aβの産生を抑えたりAβを除去したりする薬が数多く開発され、臨床試験でアルツハイマー病患者に試されてきました。しかし、そのほとんどで有効性が確認されず、開発は失敗に終わっています。この理由として、薬の投与時期が遅すぎること、薬の標的が適切でないことがあげられます。上に述べたように、アルツハイマー病を発症する頃にはすでに多くの神経細胞が死んでおり、その時点でAβを除去しても神経細胞は元に戻りません。これでは遅すぎます。また、シナプスの働きを阻害し神経細胞を死に至らしめるのは、実はAβの線維状凝集体ではなく、その前にできる目に見えない可溶性のオリゴマー(ある分子が数個~数十個集まってできた会合体)であることがわかってきました。オリゴマーこそが、シナプスの働きを阻害し、タウの過剰リン酸化を引き起こす原因だったのです。この考えは「オリゴマー仮説」と呼ばれ、2002年に提唱されました。今では、Aβばかりでなく、タウでもオリゴマーが悪いと考えられ始めています(図2)。従って、薬の標的とすべきはAβオリゴマーやタウオリゴマーです。老人斑や神経原線維変化ではありません。
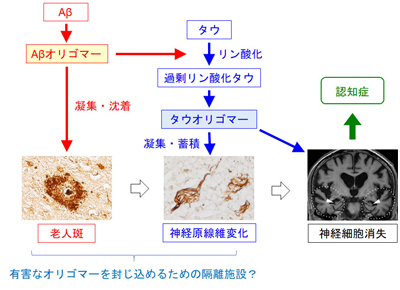
図2.アルツハイマー病の発症機序
では老人斑や神経原線維変化は何のために形成されるのでしょうか。それはおそらく、有害なオリゴマーを封じ込めるために生体が作った隔離施設のようなものだろうと考えられます。老人斑や神経原線維変化自体に毒性があるわけではありませんが、これらは、悪さをするオリゴマーが脳にあふれ始めているという目印になります。それを外から観察できれば、アルツハイマー病の早期診断に使えるでしょう。そこで、老人斑や神経原線維変化に特異的に結合し、信号を発するような試薬(プローブと呼びます)が開発されました。それを用いて老人斑や神経原線維変化を可視化したのが、アミロイド・イメージング(PIB-PETとも呼ばれます)やタウ・イメージングと呼ばれる画像診断法です。先ほど、老人斑は認知症発症の20年以上も前からできると述べましたが、これがわかったのもアミロイド・イメージングが開発されたおかげなのです。
Osaka変異発見物語
Osaka変異の発見
「アルツハイマー病は、可溶性Aβオリゴマーによるシナプス機能障害で始まる」と信じられています。しかし、このオリゴマー仮説が提唱された2002年当時は、オリゴマーによってアルツハイマー病が発症しているという直接的な証拠はありませんでした。アルツハイマー病患者の脳では目に見えない可溶性Aβと凝集して老人斑を形成した不溶性Aβとが常に混在しており、病理や臨床症状がどちらのAβによって引き起こされたものなのかを見極めることは困難でした。
2001年、ある日本人女性(当時57歳)が当大学医学部付属病院を受診しました。この方は55歳頃からもの忘れがひどくなり、受診時 軽度認知障害(MCI)と診断されました。しかし、ご家族に認知症発症者が多数いたことから、家族性アルツハイマー病が疑われました。担当医からの依頼を受けて、私たちはこの方の遺伝子診断を行い、翌2002年9月、アミロイド前駆体タンパク質(APP)遺伝子上に新しい変異を発見しました。この変異はAPPでは初めての欠失型変異で、693番目のグルタミン酸をコードするコドン(GAA)がそっくり欠失していました(図3)。この方の認知機能はその後徐々に低下し、アルツハイマー病診断基準に基づいて、59歳でアルツハイマー病と診断されました。この方のご家族の遺伝子を調べさせていただいたところ、この方、およびこの方と同じ歳に認知症を発症することになる妹さん(当時は未発症)は2つのAPP遺伝子の両方(ホモ)にこの変異を持っていました。一方、未発症のお姉さんともう一人の妹さん、および2人の娘さんは片方(ヘテロ)のAPP遺伝子にしかこの変異を持っていませんでした。この結果は、本変異(Osaka変異と命名)が家族性アルツハイマー病では非常にまれな劣性遺伝である可能性を示唆しています。
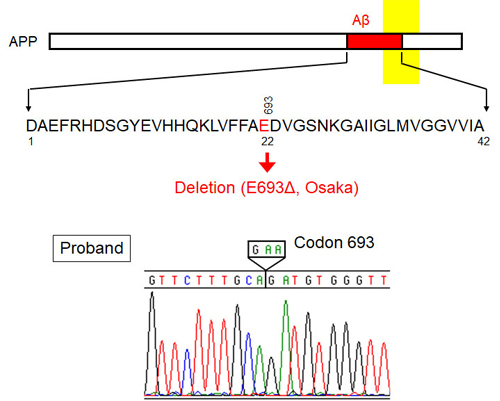
図3.Osaka変異の同定(変異の位置と患者の遺伝子診断結果)
APPの693番目のグルタミン酸は、Aβ配列内部(N末から22番目)にあります。したがって、変異APPからできてくるAβもこの変異を持っています。私たちはOsaka変異がどのようなメカニズムで病気を発症させているのかを調べました。まず、Osaka変異APP遺伝子を培養細胞に入れて発現(タンパク質を作らせること)させたところ、そこから分泌されるAβの量は野生型APPの半分近くまで減少しました。Aβ40とAβ42の比率は野生型APPとほぼ同じでした。次に、合成したOsaka変異Aβペプチドを溶液中でインキュベート(一定温度で反応させること)し、その凝集を経時的に調べました。野生型Aβは凝集してアミロイド線維(フィブリル)を作りましたが、変異Aβは100 μMの濃度で1週間インキュベートしても凝集しませんでした(図4A)。一方、変異AβをAβ分解酵素であるネプリライシンやインスリン分解酵素と反応させてみたところ、変異Aβは野生型Aβに比べ分解されにくいことがわかりました。私たちは、Osaka変異を発見してすぐに、この変異ヒトAPPを発現するトランスジェニック(Tg)マウスの作製に着手していました。2003年には発現量の異なる3つの系統が確立されました。一番発現量の多いTgマウスから脳切片を作製し、Aβに対する抗体で染めてみましたが、老人斑らしきものを見つけることはできませんでした。細胞からのAβ分泌は減少し、できたAβがフィブリルも老人斑も作らないとすれば、多少分解されにくくなっているとはいえ、この変異ははたして病気の原因なのでしょうか。むしろアルツハイマー病を発症させにくくしているのではないかと思われました。
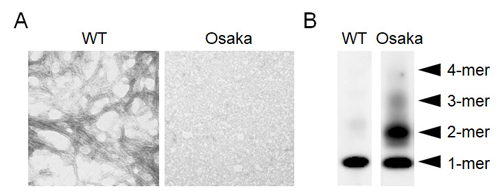
図4.Osaka変異Aβの凝集
Osaka変異の病理効果が何なのかはっきりしないまま発見から1年近くが過ぎました。2003年8月のある日、私たちは、電気泳動(サンプルをゲルにのせて電気を流し、そこに含まれる分子を分子量の大きさで分離する方法)の結果を眺めていて、ふと、変異Aβが野生型Aβよりも多くのオリゴマーを形成しているということに気づきました(図4B)。Osaka変異があると、Aβはフィブリルや老人斑を作るほどには凝集しないけれども、そこに至る前の中間産物であるオリゴマーはたくさんできている。これは全く予期せぬ性質でした。オリゴマーができたのなら、それはさらに凝集してフィブリルとなり、最終的には老人斑として脳に沈着すると考えるのが普通だったからです。このデータを見て、私たちは、Osaka変異はAβのオリゴマー化を促進し、できたオリゴマーがシナプス機能を障害することでアルツハイマー病が発症するのではないかと考えました。
私たちは早速、他大学の先生と共同で、シナプスの可塑性(この場合は神経線維の高頻度刺激によるシナプス伝達効率の変化)に対する変異Aβの効果を調べてみました。変異Aβがオリゴマーを作りやすいのなら、シナプス機能はより強く障害されるでしょう。ラットの脳室に合成Aβペプチドを注入し、10分後に神経線維を高頻度刺激して海馬での興奮性シナプス後電位(EPSP)を測定したところ、変異Aβは野生型Aβよりも強く長期増強(LTP)と呼ばれるシナプス応答を阻害しました。まさに期待通りの結果でした。
では、Osaka変異Aβは本当に老人斑を形成しないのでしょうか。このことを確認するため、2006年、私たちは、神経内科の担当医に依頼して、Osaka変異患者さんのアミロイド・イメージングを行いました。その結果、通常のアルツハイマー病患者で見られるようなシグナルは検出されませんでした(図5)。これは、この患者さんの脳には老人斑がないことを示しています。にもかかわらず、この方は認知症を発症しました。この患者さんの脳脊髄液を調べてみると、対照者に比べ、Aβの量は著しく低く、Aβモノマーに対するオリゴマーの比率が上がっていることがわかりました。脳脊髄液中のAβ量の低下は、Aβが脳実質(細胞が詰まっている空間)に溜まり、脳脊髄液に出てこなくなったからと考えられます。おそらく、脳実質に溜まったAβオリゴマーがシナプス機能を障害し、病気の引き金を引いたのでしょう。
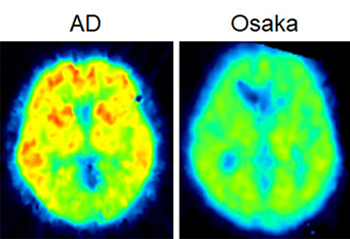
図5.Osaka変異患者のアミロイド・イメージング
2007年秋、私たちはこれまでの結果をまとめて国内および国際学会で発表しました。まだ論文になっていませんでしたので、秘密保持のため、変異の位置は伏せたままです。それでも、オリゴマーだけを作る変異が見つかったというニュースは、大きな話題となりました。さらに、「アルツハイマー病はAβオリゴマーだけで発症し、老人斑は必要ない」という私たちの発表は、「アルツハイマー病の脳には老人斑が存在する」というこれまでの常識をくつがえすもので、議論を呼び起こしました。翌2008年に、それまでリジェクト(掲載拒否)続きであった論文がようやくアクセプト(掲載可)になりました。今では、この発見はオリゴマー仮説のヒトでの証拠を示すものとして評価されています。
Osaka変異の病理作用
ところで、Osaka変異があるとなぜ細胞からのAβ分泌が低下するのでしょうか。話が前後しますが、2003年の春、私たちは、培養細胞を使った実験で、Osaka変異はAβの産生そのものには影響しないけれども、できたAβはすぐに分泌されることなく細胞内に留まったままであることを見出しました。さらに、2006年の春、Aβオリゴマーに対する抗体で細胞を染めてみると、細胞内のAβはオリゴマーを形成していることもわかりました。
これとほぼ同時期に、私たちは、Osaka変異Tgマウスで興味深い事実をつかんでいました。先ほど述べたように、このマウスの脳には老人斑はできないのですが、より詳細に調べてみると、このマウスでは8カ月齢から神経細胞内にAβが蓄積し始めていることがわかりました。オリゴマー抗体で染めてみると、ここでも、細胞内のAβはオリゴマーを形成していました(2006年2月~8月)。他大学の先生と共同で、8カ月齢のTgマウスで海馬のLTPを測定してみたところ、このマウスではLTPが低下していました(2006年2月)。モリス水迷路を用いた学習試験では、Tgマウスで顕著な記憶低下が観察されました(2006年3月)。さらに、シナプスのマーカータンパク質であるシナプトフィジンの組織染色を行ったところ、8カ月齢から加齢依存的な減少が認められました。これらの結果は、脳に溜まったAβオリゴマーがシナプスの機能を障害し、シナプス消失や認知機能低下を引き起こしたことを示しています。ちなみに、ここで述べた培養細胞での結果やTgマウスでの結果は、2007年の最初の学会発表や2008年の最初の論文からは省いています。データが多くなりすぎるとかえって話がわかりにくくなるとともに、より詳細な解析が必要となり、発表には時期尚早であるとの判断からです。
アルツハイマー病の脳では、Aβの蓄積とともに、神経原線維変化の形成、グリア細胞の活性化(脳の炎症)、ニューロンの消失など、様々な病理変化が起きています。上述したように、アルツハイマー病の脳にはオリゴマーと老人斑の両方があるため、これらの病理のうちどこまでがオリゴマーによるもので、どこからが老人斑によるものかを知ることは困難です。しかし、Osaka変異があれば、老人斑は形成されず、オリゴマーによる病理だけをみることができます。はたして、Osaka変異Tgマウスではシナプス変性以外のアルツハイマー病病理は起こっているのでしょうか。最初の論文(2008年)が発表されたのち、私たちはこのことを調べ始めました。まず、リン酸化タウに対する抗体で脳を染めたところ、TgマウスではAβオリゴマーが蓄積し始める8カ月齢で、海馬の神経線維が陽性となり、タウの病理が始まっていることが示されました。しかし、ニューロン細胞体は24カ月齢でも染まらなかったことから、神経原線維変化の形成には至ってないと考えられました。次に、アストロサイトのマーカーであるGFAPとミクログリアのマーカーであるIba-1に対する抗体を用いて脳を染めたところ、Tgマウスでは、12カ月齢より活性化ミクログリアが、18カ月齢より活性化アストロサイトが数多く検出され、脳の炎症が起こっていることが示されました。最後に、成熟ニューロンのマーカーであるNeuNに対する抗体で脳を染めたところ、24カ月齢で海馬ニューロンの有意な減少が認められました。以上の結果は、Aβオリゴマーだけで、アルツハイマー病の様々な病理が誘導されることを示しています(図6)。
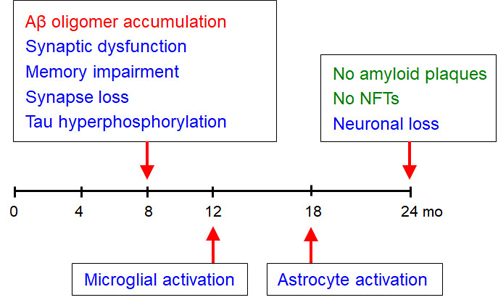
図6.Osaka変異Tgマウスの脳病理
しかし、このTgマウスでは、神経原線維変化だけはできませんでした。私たちは、これはマウスの脳だから、つまりそこにあるのがマウスのタウだからできないのかもしれない、そこにあるのがヒトのタウなら神経原線維変化はできるのではないかと考えました。そこで、野生型ヒトタウを発現するTgマウスとOsaka変異Tgマウスを掛けあわせてみたところ、このダブルTgマウスでは、18カ月齢で見事に神経原線維変化が形成されました。つまり、Aβオリゴマーさえあれば、アルツハイマー病は発症し進行すること、そこに老人斑は必要ないことがはっきり示されたのです。2014年、私たちは、他研究施設の協力のもと、Osaka変異患者さんに対して、当時実用化され始めたばかりのタウ・イメージング(神経原線維変化を検出する脳の画像診断)を行いました。結果は、この患者さんの脳には神経原線維変化が多数出現し、脳の萎縮も起こっていることが確認されました(2015年、京都で開催されたUS-Japan DIAN Collaborative Workshopで発表)。これにより、ヒトでも、Aβオリゴマーさえあれば、老人斑が無くてもアルツハイマー病は発症し進行することが決定的となりました。
Osaka変異の遺伝機序解明
始めの方で述べたように、Osaka変異は両方のAPP遺伝子に異常がなければ発症しません。すなわち、劣性遺伝です。家族性アルツハイマー病でこれまで見つかったAPPの変異はほぼすべて優性遺伝(ヘテロで発症)です。悪玉のAβオリゴマーを多く作る変異なのに、Osaka変異はなぜ優性遺伝ではないのか。この疑問は、学会発表当時から、何人かの研究者に指摘されていました。
ある変異が優性遺伝となるか劣性遺伝となるかは、次のように決まります。
A.優性遺伝となる場合
- 変異遺伝子からできる変異タンパク質が新たな機能(多くの場合、毒性)を獲得する(gain of function)。これまでに報告されているAPPの遺伝子変異はほぼすべてこれに該当します。
- 変異タンパク質が機能を喪失する(loss of function)。加えて、この変異タンパク質が、もう一方の遺伝子からできる正常なタンパク質の働きを阻害する(dominant negative effect)。
- 変異タンパク質が機能を喪失する。この変異タンパク質が悪さをするわけではないが、もう一方の遺伝子からできる正常なタンパク質だけでは量が足りず(いつもの半分量となっている)、機能を維持できない(haploinsufficiency)。
B.劣性遺伝となる場合
- 変異タンパク質は機能を喪失するが、もう一方の遺伝子からできる半分量の正常なタンパク質だけで機能を維持できる。すなわち、両方の遺伝子に変異がなければ症状は現れない。
Osaka変異はAPP遺伝子上にあり、劣性遺伝なので、APPの何らかの機能が喪失することで病気が発症していると考えられます。では、APPのどのような機能が喪失しているのでしょうか。APPの生理機能は、実はよくわかっていません。成長因子、レセプター、細胞接着因子などの可能性があげられていますが、はっきりしません。これを明らかにするには、APP遺伝子を欠失させたノックアウトマウスを作って、どんな異常が起こるかを見てみるのが一番です。相同組み換えという手法を用いてAPPノックアウトマウスが作られ、これまでにいくつかの論文が報告されています。しかし、あまり異常が起きているようには見えませんでした。
ノックアウトマウスを作るのと同じ手法で、マウスの遺伝子に変異を入れたり、マウスの遺伝子の一部をヒトの同種の遺伝子と入れ替えたりすることもできます。そのようなマウスはノックインマウスと呼ばれます。私たちは、学内の他研究室の先生と共同で、2003年にOsaka変異をマウスAPP遺伝子に挿入したノックインマウスの作製に着手していました。このマウスは、前述のTgマウスとは異なり、ヒトAβではなくマウスAβを産生します。2004年にヘテロノックインマウスができ、その雄と雌を掛け合わせてホモノックインマウスを作製しました。この新しいノックインマウスの認知機能と脳病理を調べてみると、このマウスはホモノックインでのみ、4カ月齢で記憶障害、8カ月齢でAβオリゴマーの蓄積を示すことがわかりました。つまり、マウスにおいても、Osaka変異は劣性遺伝を示したのです。しかし、Tgマウスと違い、Aβオリゴマーの蓄積と記憶障害のタイミングは一致していません。脳病理が出現する4カ月も前から認知機能がおかしくなっています。何故なのでしょう。
他研究施設の先生に依頼して、このマウスの海馬スライスを用いてシナプス機能(LTP)を測定してもらったところ、面白いことがわかりました。このLTP実験では、抑制性伝達物質GABAの働きを抑えるために、ピクロトキシンという物質を実験系に加えておきます。すると、弱い刺激でもLTPが起こりやすくなります。この条件下では、野生型マウスやヘテロノックインマウスは、4カ月齢でも8カ月齢でもLTPが誘導されました。一方、ホモノックインマウスは、4カ月齢では正常なLTPを示しましたが、8カ月齢ではLTPが抑制されていました。おそらく、8カ月齢から脳に溜まり始めたAβオリゴマーがシナプス機能を障害したのでしょう。ところが、ピクロトキシンを加えないで実験を行ってみると、妙なことが起こりました。ピクロトキシンがない条件下では、野生型マウスでもヘテロノックインマウスでもLTPは誘導されませんが、ホモノックインマウスでは4カ月齢でLTPが誘導されたのです。しかし、8カ月齢になると、やはりLTPは起こりにくくなりました。この結果が意味するところは、おそらく次のようなことです。ピクロトキシンがない状態、すなわちGABAが正常に働く状態では、GABAがシナプス活動を抑制し神経の過剰興奮を抑えるので、弱い刺激では、野生型マウスやヘテロノックインマウスではLTPは起こらない。ところがホモノックインマウスでは、おそらくGABAの働きが低下しており、弱い刺激でも神経の過剰興奮が起こり、LTPが誘導される。ただし、8カ月齢になると、Aβオリゴマーがシナプス活動を抑制するので、ホモノックインマウスでも神経の過剰興奮は起こりにくくなる。
この結果が出たちょうどその頃、私たちは、APPが海馬歯状回のGABAニューロンの維持に不可欠であることを示した論文が発表されていることに気付きました。この情報をもとに、私たちは次のように考えました。Osaka変異はAPPの機能喪失を引き起こす、その機能とは海馬GABAニューロンの維持である、従ってホモノックインマウスでは海馬のGABAニューロンが減少しているに違いない。このことを検証するため、私たちは4カ月齢のマウスの脳をGABAニューロンに対する抗体で染めてみました。すると、私たちの予想通り、ホモノックインマウスでのみ、海馬のGABAニューロンが減少していることがわかったのです。神経活動は、抑制性伝達と興奮性伝達のバランスの上に成り立っています。このバランスがくずれると、運動機能や認知機能の異常が起こります。4カ月齢のホモノックインマウスで記憶障害が観察されたのは、GABAニューロンの減少による神経の異常興奮が原因でしょう。Aβは神経活動に伴って産生分泌されるので、神経の過剰興奮が起こると、より多くのAβが産生されることになります。するとAβオリゴマーが脳に蓄積し、その毒性が発揮されるようになります。8カ月齢のホモノックインマウスでみられたAβオリゴマーの蓄積とLTPの抑制はこうしてもたらされたものでしょう。この考えが正しければ、GABAの働きを補ってさえやれば、ホモノックインマウスでも認知機能の低下は起こらずAβオリゴマーも蓄積しないことになります。そこで私たちは、GABAと同じ働きをするジアゼパムという薬を6か月齢のホモノックインマウスに2カ月間投与してみました。8カ月齢でマウスの認知機能と脳病理を調べたところ、GABAニューロンは少ないままでしたが、認知機能は正常化し、Aβオリゴマーの蓄積も起こっていませんでした。
Osaka変異が劣性遺伝なのは、ホモでこの変異を持つ場合にのみAPPの機能喪失が表面化するからでしょう。APPの機能喪失によりGABAニューロンが減少し、神経が異常興奮状態になる。するとAβ産生が増え、オリゴマーを形成し、アルツハイマー病を発症、進行させる(図7)。このように、Osaka変異で見られるAβオリゴマーの蓄積は、APPの機能喪失に伴う二次的な現象であることが、ノックインマウスを作って初めてわかりました。しかしそれでもなお、Aβオリゴマーが引き金となってアルツハイマー病の病理カスケードが進行するという事実に変わりはなく、Osaka変異はオリゴマー仮説を支持する証拠であり続けています。
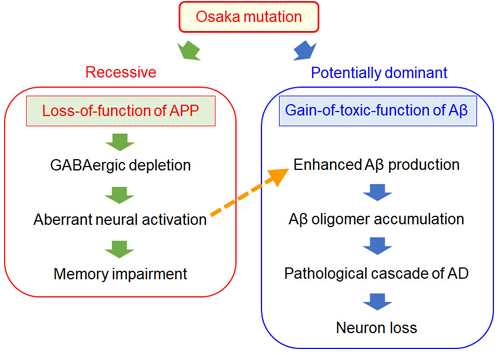
図7.Osaka変異によるアルツハイマー病発症機序
抗認知症薬の開発
- ①新しいタウ抗体の開発:
https://www.osaka-cu.ac.jp/ja/news/2014/6ieoyy
この抗体は2017年に帝人ファーマ株式会社より米メルク社に導出されました。現在はメルク社が開発を進めています。 - ②リファンピシンのドラッグ・リポジショニング:
https://www.osaka-cu.ac.jp/ja/news/2015/160329-1
リファンピシンは現在、当研究室発のベンチャーである「株式会社メディラボRFP」で開発を進めています。
株式会社メディラボRFP http://medilaborfp.com/ - ③天然物由来の認知症予防食品の開発:
これについては、当研究室発のもう一つのベンチャーである「セレブロファーマ株式会社」で開発を進めています。
セレブロファーマ株式会社 https://cerebro-p.com
Aβの生理作用の解明
- ①脳からのコレステロール排出作用:
https://onlinelibrary.wiley.com/doi/10.1002/jnr.22360
- ②末梢Aβのインスリン分泌抑制作用:
https://www.pnas.org/doi/10.1073/pnas.2117723119
メディア掲載情報
- 老人斑を作らない遺伝子変異(Osaka変異)の発見
2007年11月19日 Alzheimer Research Forumウェブサイト 2008年2月27日 朝日新聞、日本経済新聞、産経新聞、日経産業新聞、
日経バイオテクONLINE2008年2月28日 読売新聞 2008年3月16日 毎日新聞 下山 進 著「アルツハイマー征服」(2021, 角川書店)でも紹介されました (第16章)。
- Aβオリゴマーモデルマウスの開発
2010年4月8日 NHK「おはよう関西」
朝日新聞、読売新聞、日本経済新聞、日経産業新聞2010年4月9日 Alzheimer Research Forumウェブサイト 2010年4月13日 毎日新聞 - 新しいタウ抗体の開発
2015年1月9日 NHK「ニューステラス関西」「関西845」「ニュースウォッチ9」「BSニュース」関西テレビ「ニュースJAPAN」
産経WESTウェブサイト2015年1月10日 日本経済新聞、読売新聞、毎日新聞 2015年1月15日 日刊工業新聞 2015年2月10日 朝日新聞 - リファンピシンの認知症予防作用の発見
- 天然物由来の認知症予防食品の開発
2022年1月27日 Lab BRAINS「食品で実現する認知症予防」 - Aβの生理作用の解明
2022年9月12日 朝日新聞(夕刊)「Aβ」二つの病気結ぶカギ
| 2016年3月29日 | NHK 「関西のニュース」 朝日新聞(夕刊)、毎日新聞(夕刊)、産経新聞(夕刊) |
| 2016年3月30日 | 日刊工業新聞 |
| 2016年4月4日 | 読売新聞(夕刊) |
| 2016年4月25日 | 日経バイオテク4月25日号「研究室探訪」 |
| 2018年2月4日 | 日本経済新聞電子版「安すぎて開発できず アルツハイマー病治療薬の苦悩」 |
| 2018年9月9日 | 日本経済新聞「アルツハイマー 治療より予防」 |
| 2019年2月21日 | 週刊新潮「認知症1000万人時代に朗報!「アルツハイマー」予防に劇的効果の既存薬」日経バイオテク4月25日号「研究室探訪」 |
| 2019年5月2日 | テレビ朝日「羽鳥慎一モーニングショー そもそも総研『そもそも既存薬を使ってアルツハイマー病を予防できるって本当?』」 |
| 2021年12月10日 | Lab BRAINS「健康寿命100歳に向けた認知症予防とは」 |
| 2022年1月12日 | JST Science Portal「アルツハイマーなど認知症予防の点鼻薬、既存2成分で効果」 |
| 2022年1月18日 | 「MedPeer」内「MEDICAL NEWS LINE」「2つの有効成分による点鼻薬で認知症予防」 |
| 2022年1月19日 | 「健達ねっと」【専門家インタビュー】「認知症予防薬・予防食品の開発を通して認知症のない社会を目指す」 |
| 2022年1月19日 | テレビ大阪「やさしいニュース」「認知症に点鼻薬 進む治療薬の開発」 |
| 2023年1月31日 | 日経バイオテク「経鼻リファンピシンでアルツハイマー病の進行阻止へ」 |
| 2023年3月24日 | 日経バイオテク「メディラボRFP、経鼻リファンピシンが米国でオーファン指定」 |